Getting Started with NMFscape
Michael Totty
2025-09-12
Source:vignettes/getting-started.Rmd
getting-started.RmdIntroduction
NMFscape provides fast, scalable non-negative matrix factorization (NMF) for single-cell and spatial genomics data. NMF decomposes gene expression data into biologically interpretable gene expression programs and their usage patterns across cells.
Why NMF?
- Biologically interpretable: Gene programs contain only positive weights
-
Parts-based decomposition: Identifies co-expressed
gene modules
- Cell type discovery: Programs often correspond to cell type signatures
- Additive mixing: Cells can express multiple programs simultaneously
NMFscape uses RcppML for high-performance computation and integrates seamlessly with Bioconductor workflows.
library(NMFscape)
#> Loading required package: BiocGenerics
#> Loading required package: generics
#>
#> Attaching package: 'generics'
#> The following objects are masked from 'package:base':
#>
#> as.difftime, as.factor, as.ordered, intersect, is.element, setdiff,
#> setequal, union
#>
#> Attaching package: 'BiocGenerics'
#> The following objects are masked from 'package:stats':
#>
#> IQR, mad, sd, var, xtabs
#> The following objects are masked from 'package:base':
#>
#> anyDuplicated, aperm, append, as.data.frame, basename, cbind,
#> colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
#> get, grep, grepl, is.unsorted, lapply, Map, mapply, match, mget,
#> order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
#> rbind, Reduce, rownames, sapply, saveRDS, table, tapply, unique,
#> unsplit, which.max, which.min
#> Loading required package: SingleCellExperiment
#> Loading required package: SummarizedExperiment
#> Loading required package: MatrixGenerics
#> Loading required package: matrixStats
#>
#> Attaching package: 'MatrixGenerics'
#> The following objects are masked from 'package:matrixStats':
#>
#> colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
#> colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
#> colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
#> colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
#> colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
#> colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
#> colWeightedMeans, colWeightedMedians, colWeightedSds,
#> colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
#> rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
#> rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
#> rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
#> rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
#> rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
#> rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
#> rowWeightedSds, rowWeightedVars
#> Loading required package: GenomicRanges
#> Loading required package: stats4
#> Loading required package: S4Vectors
#>
#> Attaching package: 'S4Vectors'
#> The following object is masked from 'package:utils':
#>
#> findMatches
#> The following objects are masked from 'package:base':
#>
#> expand.grid, I, unname
#> Loading required package: IRanges
#> Loading required package: GenomeInfoDb
#> Loading required package: Biobase
#> Welcome to Bioconductor
#>
#> Vignettes contain introductory material; view with
#> 'browseVignettes()'. To cite Bioconductor, see
#> 'citation("Biobase")', and for packages 'citation("pkgname")'.
#>
#> Attaching package: 'Biobase'
#> The following object is masked from 'package:MatrixGenerics':
#>
#> rowMedians
#> The following objects are masked from 'package:matrixStats':
#>
#> anyMissing, rowMedians
#> Warning: replacing previous import 'BiocGenerics::sd' by 'stats::sd' when
#> loading 'NMFscape'
library(SingleCellExperiment)
library(scuttle)
library(scater)
#> Loading required package: ggplot2
library(scran)
library(scRNAseq)
library(ggplot2)
library(patchwork)Basic NMF Workflow
Load and prepare data
We’ll use the Zeisel brain dataset (Zeisel et al. 2015, Science) which contains diverse brain cell types.
# Load brain dataset
sce <- ZeiselBrainData()
cat("Original data:", nrow(sce), "genes,", ncol(sce), "cells\n")
#> Original data: 20006 genes, 3005 cells
# Quality control and filtering
sce <- addPerCellQCMetrics(sce)
sce <- addPerFeatureQCMetrics(sce)
sce <- sce[rowData(sce)$detected >= 10, ] # Genes detected in ≥10 cells
sce <- sce[, colData(sce)$detected >= 500] # Cells with ≥500 detected genes
# Subset to 1000 cells for computational efficiency
set.seed(42)
if (ncol(sce) > 1000) {
cell_subset <- sample(ncol(sce), 1000)
sce <- sce[, cell_subset]
}
# Normalize and select highly variable genes
sce <- logNormCounts(sce)
dec <- modelGeneVar(sce)
top_hvgs <- getTopHVGs(dec, n = 1000) # Top 1000 HVGs
sce <- sce[top_hvgs, ]
cat("Final data:", nrow(sce), "genes,", ncol(sce), "cells\n")
#> Final data: 1000 genes, 1000 cellsRun NMF
# Run NMF with 8 programs (good for brain cell diversity)
sce <- runNMFscape(sce, k = 8, verbose = FALSE)
#>
#> Attaching package: 'Matrix'
#> The following object is masked from 'package:S4Vectors':
#>
#> expand
# Check results
cat("NMF programs stored in:", reducedDimNames(sce), "\n")
#> NMF programs stored in: NMF
cat("Basis matrix stored in:", names(metadata(sce)), "\n")
#> Basis matrix stored in: NMF_basisAdd dimension reductions for visualization
# Run PCA and UMAP for visualization
sce <- runPCA(sce, ncomponents = 50)
sce <- runUMAP(sce, dimred = "PCA")
cat("Available dimension reductions:", reducedDimNames(sce), "\n")
#> Available dimension reductions: NMF PCA UMAPVisualizing NMF Results
Plot NMF programs on UMAP
NMFscape provides convenient visualization functions to plot NMF program usage on dimension reduction coordinates.
# Visualize two distinct NMF programs
p1 <- vizUMAP(sce, program = 1, title = "NMF Program 1")
p2 <- vizUMAP(sce, program = 3, title = "NMF Program 3", color_scale = "plasma")
# Display side by side
p1 + p2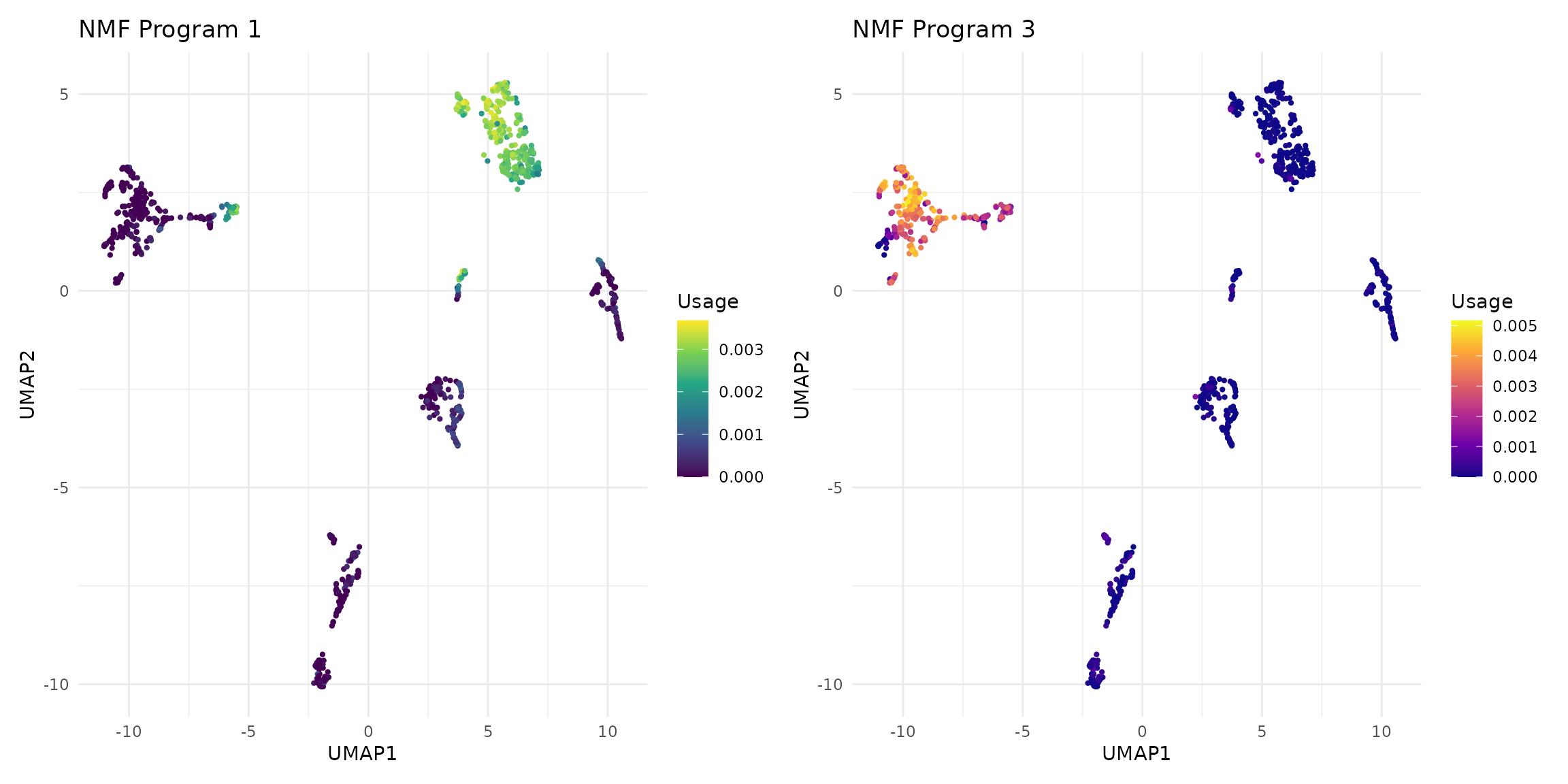
# Compare with known cell types
p3 <- plotReducedDim(sce, dimred = "UMAP", colour_by = "level1class",
point_size = 0.8) +
ggtitle("Known Cell Types") +
theme(legend.position = "right")
# Show NMF programs vs cell types
(p1 + p2) / p3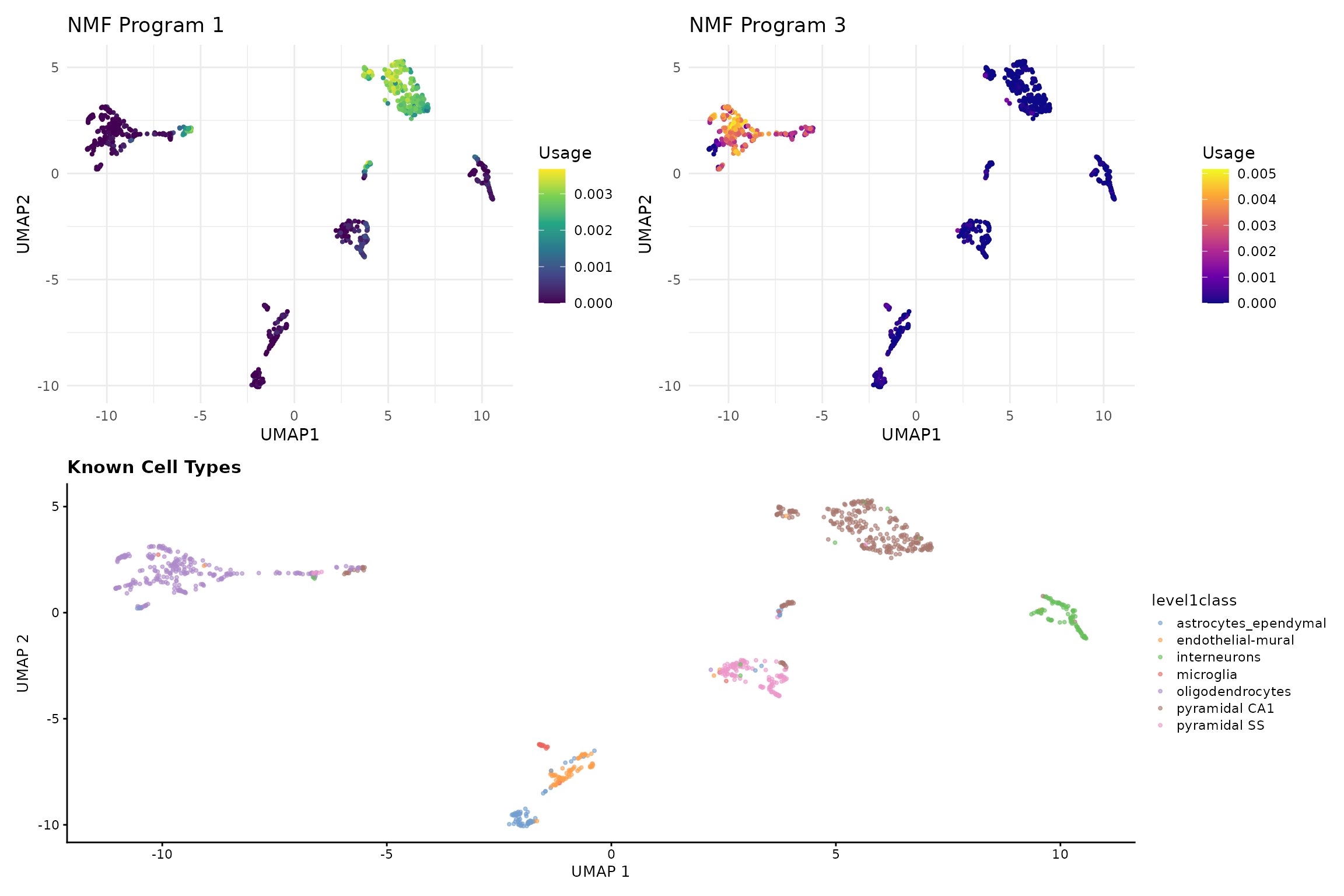
Examine top genes per program
# Get top contributing genes for each program
top_genes <- getTopFeatures(sce, n = 10)
# Display top genes for first few programs
for (i in 1:4) {
cat("Program", i, "top genes:", paste(top_genes[[i]][1:5], collapse = ", "), "...\n")
}
#> Program 1 top genes: Atp2b1, Meg3, Gria2, Ppp3ca, Atp1b1 ...
#> Program 2 top genes: Usmg5, Mdh1, Calm2, Cycs, Atpif1 ...
#> Program 3 top genes: Plp1, Trf, Apod, Mal, Car2 ...
#> Program 4 top genes: Meg3, Nrgn, Arpp21, Mef2c, Atp2b1 ...Key Functions
-
runNMFscape(): Run NMF decomposition -
getBasis(): Extract gene expression programs (genes × programs) -
getCoefficients(): Extract program usage (cells × programs)
-
getTopFeatures(): Get top genes per program -
vizUMAP(): Visualize programs on UMAP coordinates -
vizDimRed(): Visualize programs on any dimension reduction
Session Information
sessionInfo()
#> R version 4.5.1 (2025-06-13)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.3 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] Matrix_1.7-3 patchwork_1.3.2
#> [3] scRNAseq_2.22.0 scran_1.36.0
#> [5] scater_1.36.0 ggplot2_4.0.0
#> [7] scuttle_1.18.0 NMFscape_0.99.0
#> [9] SingleCellExperiment_1.30.1 SummarizedExperiment_1.38.1
#> [11] Biobase_2.68.0 GenomicRanges_1.60.0
#> [13] GenomeInfoDb_1.44.2 IRanges_2.42.0
#> [15] S4Vectors_0.46.0 MatrixGenerics_1.20.0
#> [17] matrixStats_1.5.0 BiocGenerics_0.54.0
#> [19] generics_0.1.4 BiocStyle_2.36.0
#>
#> loaded via a namespace (and not attached):
#> [1] RColorBrewer_1.1-3 jsonlite_2.0.0 magrittr_2.0.3
#> [4] gypsum_1.4.0 ggbeeswarm_0.7.2 GenomicFeatures_1.60.0
#> [7] farver_2.1.2 rmarkdown_2.29 BiocIO_1.18.0
#> [10] fs_1.6.6 ragg_1.5.0 vctrs_0.6.5
#> [13] Rsamtools_2.24.1 memoise_2.0.1 RCurl_1.98-1.17
#> [16] htmltools_0.5.8.1 S4Arrays_1.8.1 AnnotationHub_3.16.1
#> [19] curl_7.0.0 BiocNeighbors_2.2.0 Rhdf5lib_1.30.0
#> [22] rhdf5_2.52.1 SparseArray_1.8.1 alabaster.base_1.8.1
#> [25] sass_0.4.10 bslib_0.9.0 alabaster.sce_1.8.0
#> [28] desc_1.4.3 httr2_1.2.1 cachem_1.1.0
#> [31] GenomicAlignments_1.44.0 igraph_2.1.4 lifecycle_1.0.4
#> [34] pkgconfig_2.0.3 rsvd_1.0.5 R6_2.6.1
#> [37] fastmap_1.2.0 GenomeInfoDbData_1.2.14 digest_0.6.37
#> [40] AnnotationDbi_1.70.0 dqrng_0.4.1 irlba_2.3.5.1
#> [43] ExperimentHub_2.16.1 textshaping_1.0.3 RSQLite_2.4.3
#> [46] beachmat_2.24.0 labeling_0.4.3 filelock_1.0.3
#> [49] httr_1.4.7 abind_1.4-8 compiler_4.5.1
#> [52] bit64_4.6.0-1 withr_3.0.2 S7_0.2.0
#> [55] BiocParallel_1.42.1 viridis_0.6.5 DBI_1.2.3
#> [58] alabaster.ranges_1.8.0 HDF5Array_1.36.0 alabaster.schemas_1.8.0
#> [61] rappdirs_0.3.3 DelayedArray_0.34.1 rjson_0.2.23
#> [64] bluster_1.18.0 tools_4.5.1 vipor_0.4.7
#> [67] beeswarm_0.4.0 glue_1.8.0 h5mread_1.0.1
#> [70] restfulr_0.0.16 rhdf5filters_1.20.0 grid_4.5.1
#> [73] cluster_2.1.8.1 gtable_0.3.6 ensembldb_2.32.0
#> [76] RcppML_0.3.7 BiocSingular_1.24.0 ScaledMatrix_1.16.0
#> [79] metapod_1.16.0 XVector_0.48.0 ggrepel_0.9.6
#> [82] BiocVersion_3.21.1 pillar_1.11.0 limma_3.64.3
#> [85] dplyr_1.1.4 BiocFileCache_2.16.2 lattice_0.22-7
#> [88] FNN_1.1.4.1 rtracklayer_1.68.0 bit_4.6.0
#> [91] tidyselect_1.2.1 locfit_1.5-9.12 Biostrings_2.76.0
#> [94] knitr_1.50 gridExtra_2.3 bookdown_0.44
#> [97] ProtGenerics_1.40.0 edgeR_4.6.3 xfun_0.53
#> [100] statmod_1.5.0 pheatmap_1.0.13 UCSC.utils_1.4.0
#> [103] lazyeval_0.2.2 yaml_2.3.10 evaluate_1.0.5
#> [106] codetools_0.2-20 tibble_3.3.0 alabaster.matrix_1.8.0
#> [109] BiocManager_1.30.26 cli_3.6.5 uwot_0.2.3
#> [112] systemfonts_1.2.3 jquerylib_0.1.4 Rcpp_1.1.0
#> [115] dbplyr_2.5.1 png_0.1-8 XML_3.99-0.19
#> [118] parallel_4.5.1 pkgdown_2.1.3 blob_1.2.4
#> [121] AnnotationFilter_1.32.0 bitops_1.0-9 alabaster.se_1.8.0
#> [124] viridisLite_0.4.2 scales_1.4.0 crayon_1.5.3
#> [127] rlang_1.1.6 cowplot_1.2.0 KEGGREST_1.48.1